Per Poem@Home:
Computational Alanine Screen 1ILP CXCR1 project
Chemokines are small soluble chemotactic proteins that are recognized by chemokine receptors on immune cells and other cell types. They direct these cells to their site of action in inflammation or development.
Due to their role in chronic inflammation, allergies, autoimmune diseases and cancer, chemokines have become a target in drug discovery. Starting from existing structural data (PDB:
1ILP) of a chemokine interleukin-8
CXCL8 bound to a N-terminal peptide of its corresponding receptor CXCR1. We used the all-atom free-energy PFF02 on POEM@HOME to calculate the interaction energy differences between the wild-type complex and all alanine-exchanged peptides to determine the interaction hot spots. These changes are rationalized by decomposing the interaction energy differences into ther most impotant physical contributions.
Structure of the Interleukin dimer bound to the 19 N-terminal amino acids of its cognate receptor CXCR1. The N-terminal methionine was exchanged for alanine.
The results of our analysis agree with available experimental functional assays, indicating that this approach is suitable for computational alanine screening and may help to identify competitive peptides as starting points for the development of inhibitors of protein-protein interactions for pharmaceutically relevant targets.
The following two graphs show the results for the docking simulations carried out on POEM@HOME.
Energy vs. RMSD plot of all relaxed conformations of 1ILP yields two candidate conformations with nearly identical energy. The chemokine is shown in blue, while the competing peptide conformations are shown in red and magenta.
Further relaxation of the lowest 60 conformations uniquely identified the native model to within 0.5 Angstroem. The chemokine is shown in blue, the native model in red and the docked model as spheres.
These results were published in the Journal of Chemical Physics: Probing hot spots on protein-protein interfaces with all-atom free-energy simulation Irene Meliciani, Konstantin Klenin, Timo Strunk, Katja Schmitz, and Wolfgang Wenzel, J. Chem. Phys. 131, 034114 (2009), DOI:10.1063/1.3177008
A link to this article is for example
here. If you do not have access to this article, very often libraries provide access free-of-charge.
Anche se il progetto è in fase alpha è un risultato molto interessante.
P.S.: alleluiah, siamo di nuovo up.... per quanto?


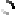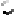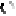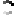



Originariamente Scritto da SaTaN SHaRK












